Biomolecular NMR and Metabolomics for disease metabotyping
Key Research Interests
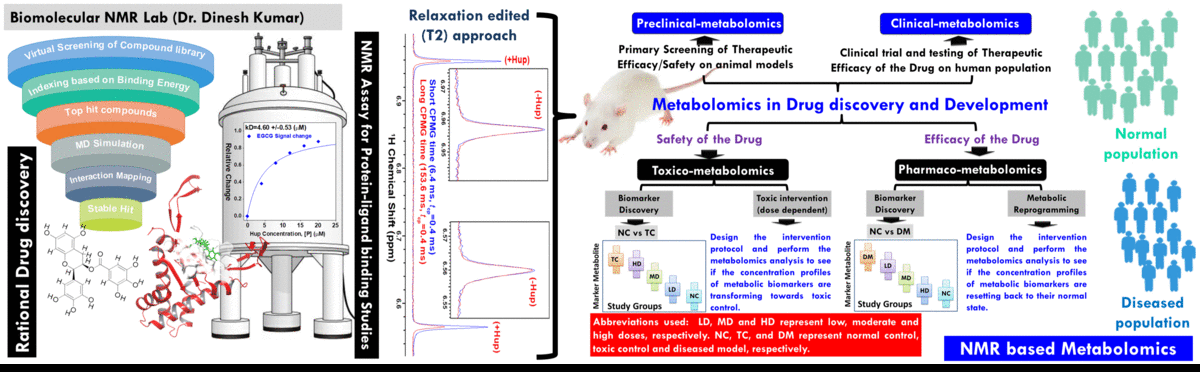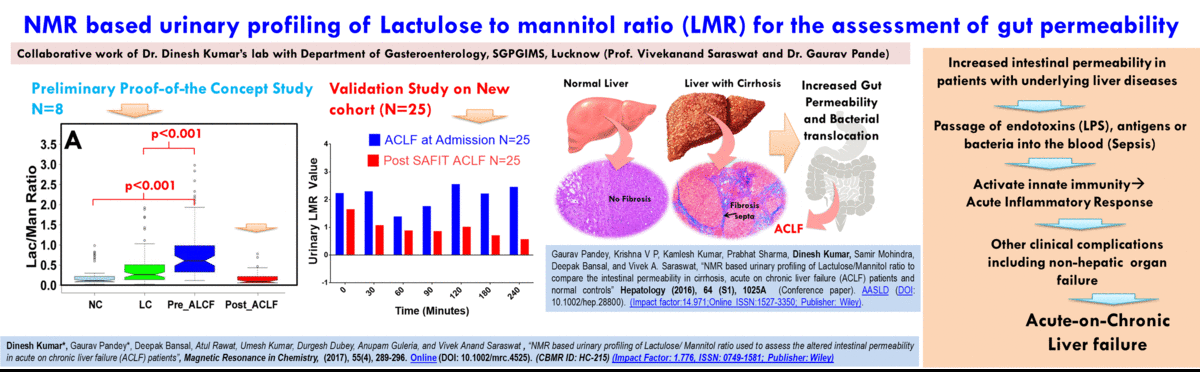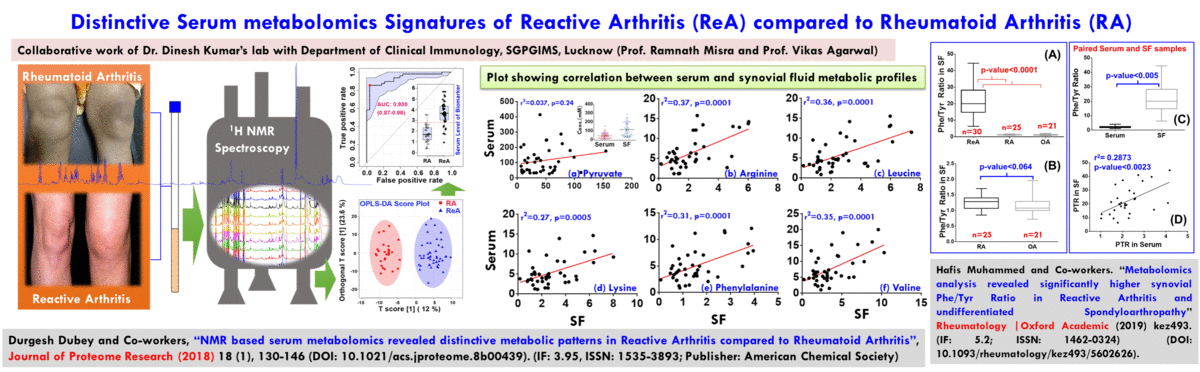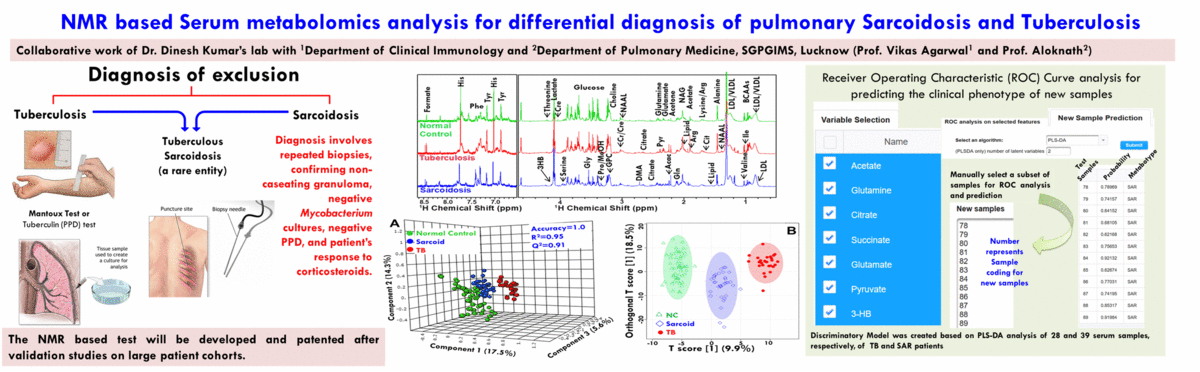
- The broad area of my research at CBMR is Biomolecular NMR and primarily we are using this for clinical and preclinical metabolomics studies related to (a) disease metabotyping i.e. to identify the disease specific metabolic signatures and to explore their utility in clinical diagnosis and surveillance and (ii) evaluation of efficacy and safety of potential therapeutic candidates including traditional herbal medicines.
- In addition to metabolomics studies, we are also involved in developing novel NMR methodologies for high throughput structural and functional studies of proteins and further employing these methods for studying mechanistic structural biology of proteins of therapeutic relevance. The methodological developments are aimed (a) to facilitate the protein structure determination process, (b) rapid characterization of protein conformation features and (c) studying their interactions with their physiological binding partners (like peptides/proteins, lipids, carbohydrates, nucleic acids, metal ions, etc) or rationally designed small molecule inhibitors (including small synthetic molecules or natural products) to validate structure-activity relations (SAR) by NMR. Particularly, the rapid methods are highly desirable for structural investigations of proteins which are either unstable in solution or tend to precipitate in matter of days.
- In parallel, we are employing the NMR methods for studying the Mechanistic Structural Biology of Histone like DNA binding Proteins (HU) of formidable pathogenic bacteria (like Mycobacterium Tuberculosis, Helicobacter pylori or Streptococcus pneumoniae). The resulted structural information would be subsequently used to guide the rational discovery of their inhibitors as starting precursors for developing next generation antibiotics.
Research Overview:
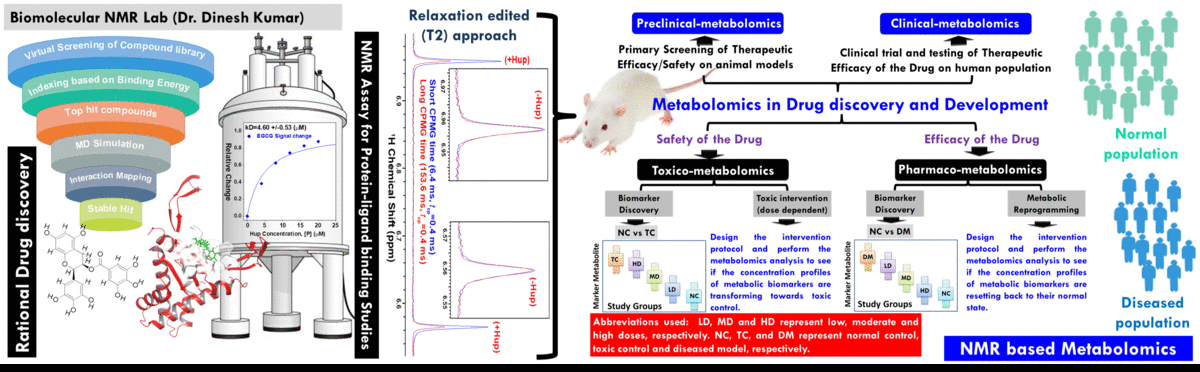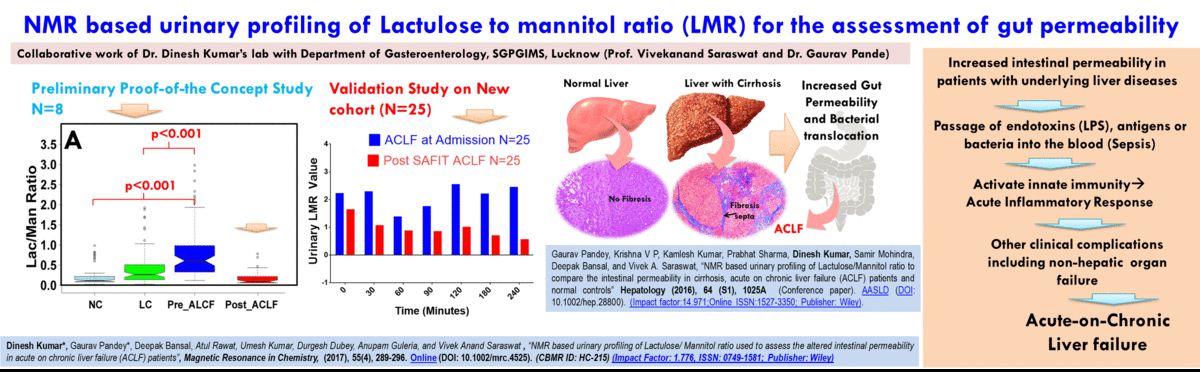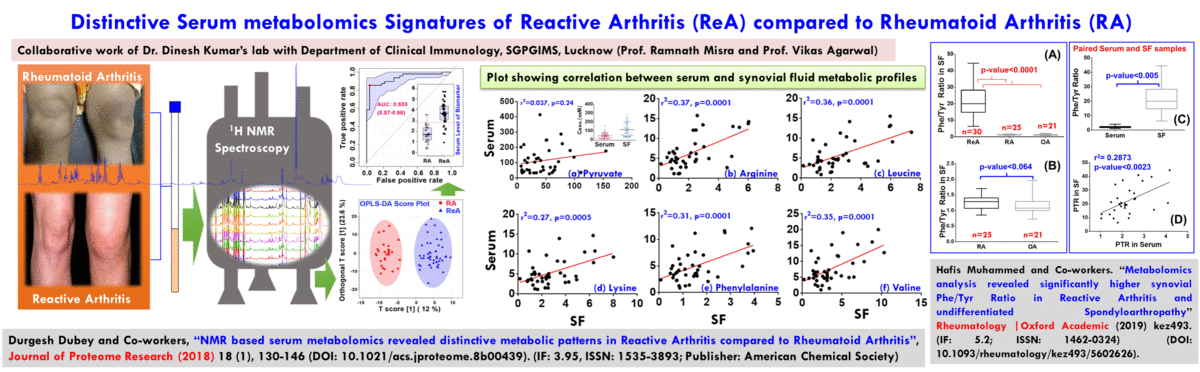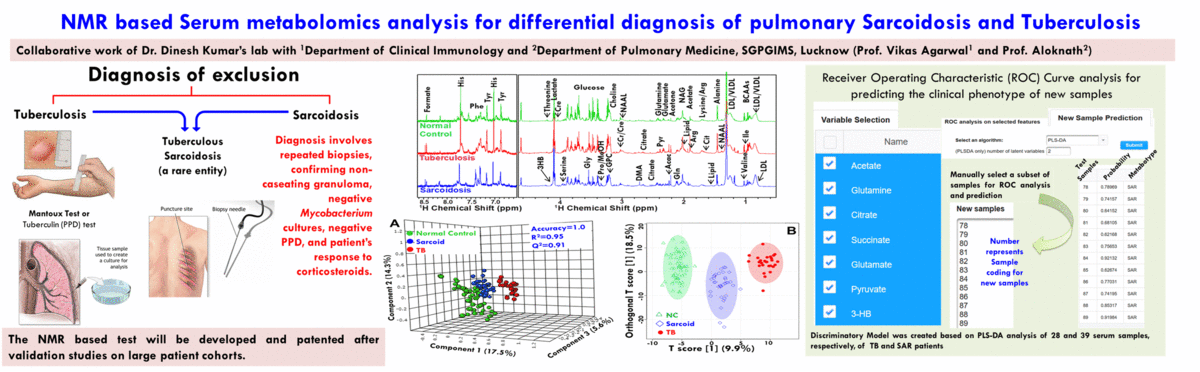
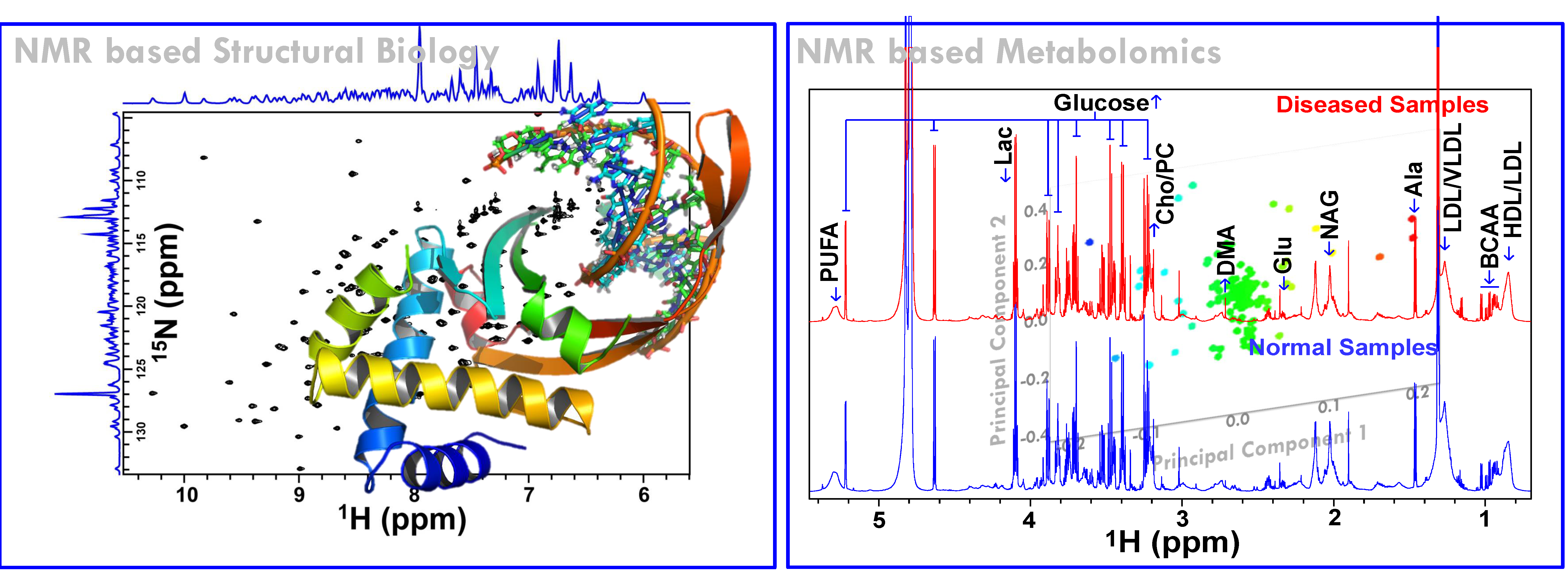
(A) NMR based Metabolomics: Metabolomics is a promising approach to reveal altered metabolism induced by a disease or its therapeutic intervention. Therefore, it is gradually becoming a mutually complementary technique to genomics, transcriptomics and proteomics both for identifying disease specific biomarkers and evaluating the efficacy and safety of pharmaceutical products. NMR coupled with multivariate analysis is currently the method of choice for rapid metabolomics analysis owing to its unbiased, non-destructive nature and minimal sample preparation requirement. Known as NMR based metabolomics has been extensively and exclusively used in our group to identify the metabolic signatures of various rheumatic diseases (including Rheumatoid arthritis, reactive arthritis, Osteoarthritis, Takayasu Arteritis, Lupus, Lupus nephritis, and small vessel vasculitis) and other devastating human diseases like Acute-on-chronic liver failure and acute myocardial infarction.
(B) NMR based Protein structural Biology: Proteins are bio-nano-machines (or so called biology’s workhorses) designed by molecular evolution for various cellular activities (Peng J W, Structure 2009; (17): 319-320). The elucidation of their structure and dynamics features is the key to understand the underlying mechanisms of action of these bio-nano-machines and so to understand complete metabolic pathways, biological processes, and eventually entire organisms. This has led to the origin of a new area in biology named “Structural proteomics -the high-throughput endeavor of solving three-dimensional protein structures encoded in an organism’s genome”. In recent years, structural proteomics has found its great implication in drug industry to guide the drug discovery process, this is because when a protein is implicated in a certain disease (such as that of the SARS Coronavirus or Swine flu virus), its three dimensional structure provides the information to design a drug to interfere with the action of that protein (the process is commonly known as Structure based drug designing). Basically a drug is designed to block the active site of the protein involved in disease to suppress its harmful activity.
Among the various techniques used for structural and functional proteomics studies, NMR has emerged as the most powerful one, especially because of its ability to provide atomic level information about protein structure and dynamics. Additionally, NMR is unparalleled in its ability to unravel detailed structural and dynamical information of unfolded/denatured and flexible proteins and can also be used to determine low-resolution structures of target-ligand complexes for natively unstructured proteins (such studies are not at all amenable to X-ray crystallographic approaches). However, NMR suffers only in terms of its throughput because of the long experiment time requirement to record several multidimensional NMR experiments for structure determination. The long experiment time requirement not only limits the utility of NMR for high-throughput structural proteomics studies, but it also imposes a long time stability condition on the protein samples. However, many proteins (especially the eukaryotic/mammalian proteins) in solution tend to precipitate in a matter of days, thereby reducing the time available to record NMR data. Thus, there is very high demand for (i) reducing the number of NMR experiments to obtain required information, (ii) to increase the speed of data collection, and (iii) to develop high throughput procedures and related algorithms/techniques for fast data analysis for resonance assignment and structure determination.

The whole protein world can be explored by NMR
Working in this direction, novel NMR methods and protocols have been designed by tweaking previously described HNN and HN(C)N experiments (Panchal et al., J Biomol NMR 2001; (20): 135-147) and integrating them with other novel and fast NMR methods for speeding up protein structure determination process both in terms of data acquisition and data analysis. To start with, we modified the HNN and HN(C)N experiments for the generation of Serine/threonine check points followed by development of simple and swift protocols (based on variants of HNN and HNCN experiments) for efficient and unambiguous sequence specific assignment of backbone resonances of folded and unfolded proteins [1, 7]. Next, we modified these two remarkable pulse sequences according to BEST (Band-selective Excitation (Optimized Flip-angle) Short-Transient) NMR approach (Schanda et al., J Am Chem Soc 2006; (128): 9042-9043) for fast data collection. The modifications, named here as BEST-HNN and BEST-HN(C)N [8], have been resulted in significant reduction in total experiment time without any loss in sensitivity and resolution. Next, we have developed a simple and swift strategy (followed by its automation) based on the orthogonal projection planes of these two experiments HNN/HN(C)N for establishing the sequence specific backbone assignment of small well folded proteins in less than a day [10,11,14]. Overall, these NMR experiments will be of immense value for unambiguous and rapid resonance assignments of proteins, particularly in the context of high-throughput structural proteomics. The various developments are outlined here:

HNN/HN(C)N: Provide Direct sequential Amide correlations. These experiments have three variants: glycine variant, alanine variant, and serine/threonine Variant. Depending upon the variant of the experiment, these provide identification of certain specific triplet stretches containing glycines, alanines, and serines/threonines.
HNN-ST/HN(C)N-ST: Modified HNN/HN(C)N experiments for the generation of serine and threonine check points in their respective 3D Spectra.
BEST_HNN/BEST_HN(C)N: HNN/HN(C)N experiments (modified according to BEST NMR approach) for rapid data acquisition.
hNCOcanH: An alternative complementary experiment for HNN. Provide Direct Hit to the sequential (i-1) HSQC peak in combination with HNN. Like HNN, the experiment has three variants. Depending upon the amino-acid sequence, the variants of HNN and NCOH can be selected for complete sequence specific backbone assignment.
hnCOcaNH: The experiment leads to a spectrum equivalent to HNCACO but it provides direct distinction of self and sequential peaks as they appear opposite in sign. Thus the requirement of complementary experiment (i.e. HNCO) can be avoided.
hncoCANH: The experiment leads to a spectrum equivalent to HNCA but it provides direct distinction of self and sequential peaks as they appear opposite in sign. Thus the requirement of complementary experiment (i.e. HNCOCA) can be avoided.
2D-(HN)NH: Provides amino acid type identification of glycines, alanines and serines/threonines directly on the HSQC type spectrum.
2D-hncNH: Provides amino acid type identification of glycines, alanines, serines/threonines and the residues following them in the sequence directly on the HSQC type spectrum.
2D-hNcnH: Provides intra- and inter-residue amide correlations directly on the HSQC type spectrum.
2D-hncoCAnH: Provides intra- and inter-residue 13Ca correlations directly on the HSQC type spectrum.
2D-hnCOcanH: Provides intra- and inter-residue 13C’ correlations directly on the HSQC type spectrum.
hNCAnH: Provides 13Ca filtering of sequentially connected HSQC peaks in 13C/15N labeled proteins (for folded proteins).
hNcoCAnH/hNCAnH: An efficient high-throughput strategy -named <direct sequential hit> strategy- based on the complementary and mutually exclusive information contained in
these two spectra has been presented for unambiguous and accurate backbone assignment, especially of intrinsically unfolded proteins. The main strength of the strategy is that it provides directly the coordinates of sequential amide peaks (Hi−1, Ni−1 and Hi+1, Ni+1) for a particular amide cross peak (Hi, Ni), just after performing the simple manual peak picking step. Thus, the sequential (i to i −1 or i to i +1) connectivities between the backbone amide cross peaks (Hi, Ni) are established directly without maki
ng cumbersome search through various planes of the 3D spectrum as is the case with other presently used approaches for backbone assignment.
AUTOBA: AUTOmated Backbone Assignment based on Sequential Correlations orthogonal Projection planes of HNN and HN(C)N suite of experiments.
PFBD: Recently, an efficient semi-automated strategy called PFBD (i.e. Protein Fold from Backbone Data only) has been designed for rapid backbone fold determination of small proteins. The strategy make
s use of backbone NMR data only.
Reduced Dimensionality Tailored TOCSY/NOESY Experiments: Continuing our efforts towards high quality protein structure determination by NMR, a series of five (15N ± 13C’) edited TOCSY and NOESY experiments have been designed for Unambiguous side chain and NOE assignments of Proteins with High Shift Degeneracy.
Reduced Dimensionality Tailored HN(C)N Experiments e.g. (3,2)D-hNCOcanH and (3,2)D-hNcoCAnH: To enhance the utility of PFBD approach for medium sized proteins.
Reduced Dimensionality Tailored HN(C)N Experiments e.g. (4,3)D-hnCOCANH, (4,3)D-hNCOcaNH and (4,3)D-hNcoCANH: To enhance the utility of HN(C)N based Assignment Protocol for proteins exhibiting highly crowded HSQC spectrum.

The range of applications of the assignment method based on the single reduced dimensionality (4,3)D- hnCOCANH experiment.
Pseudo 5D HN(C)N -also referred as (5,3)D-hNCO–CANH: To extend the utility of HN(C)N based Assignment Protocol for proteins exhibiting high degree of amide and carbon shift degeneracy.
Taken together, these experiments and based strategies have their implications in NMR structural investigations of proteins.
The other interest of our lab is to explore the mechanistic structural biology of proteins of therapeutic relevance. Proteins are the most versatile bio-molecules and are involved in various important cellular and biological activities as shown here. In last two decades, Biomolecular NMR has flourished dramatically and has emerged out as a powerful tool for (a) three-dimensional (3D) structure determination of proteins, (b) screening of functional protein-protein, protein-nucleic acid and protein-ligand interactions, (c) characterization of their dynamics features at atomic level, all critically important to understand “how do these proteins function and how can their activity be altered?”. This is what which is exploited in modern drug discovery programs today where the basic aim is “to inhibit the functioning of the metabolic pathway responsible for the diseased state by causing a key molecule to stop functioning”. And what are these key molecules; the answer is Proteins. Because these are the proteins -which either in the form of enzymes, receptors, or transporters etc.- are involved in almost every cellular/metabolic activity within the cell. Studies have clearly shown that proteins use internal motions for binding, catalysis, and signal transduction. To get detailed insights into conformational dynamics on multiple time scales, we generally complement the information obtained from 15N NMR relaxation measurements with those from molecular dynamics (MD) simulations. This is because of the fact that the timescales probed using molecular dynamics (MD) simulations differ from those accessible by NMR spectroscopy, giving a more complete picture of the backbone dynamics. Molecular dynamics (MD) simulations can also be used to probe the molecular interactions (through docking) and to investigate the structure-activity relationships.
Proteins the workhorses of the Cell

